Hydroborowanie
Hydroborowanie, borowodorowanie, reakcja Browna – reakcja chemiczna, w której borowodór (często diboran, B
2H
6) reaguje z alkenem tworząc związek boroorganiczny (alkiloboran)[1][2]. Alkiloborany są uniwersalnymi reagentami do syntez organicznych.
Odkrycie i zastosowanie
edytujReakcja ta zaproponowana została w 1957 roku przez zespół badawczy prof. Herberta C. Browna[3]. W 1979 roku Brown otrzymał za to odkrycie Nagrodę Nobla w dziedzinie chemii[4][5]. Pierwszym zastosowaniem hydroborowania była synteza alkoholi z alkenów poprzez dwuetapową reakcję hydroborowania z utlenianiem. Później opracowano również wykorzystanie reakcji hydroborowania do syntezy alkilopodstawionych pochodnych α-bromoketonów oraz α-bromoestrów kwasów karboksylowych. Reakcja opisana po raz pierwszy przez Browna dotyczyła syntezy 1-heksanolu z 1-heksenu według równania[3]:
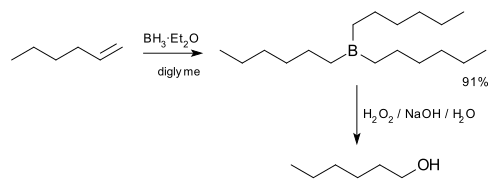
- Synteza 1-heksanolu z 1-heksenu (H.C. Brown 1975)

Hipotetyczna struktura najprostszego borowodoru (BH
3) jest nietrwała, dlatego w reakcji tej stosuje się jako rozpuszczalniki etery takie jak tetrahydrofuran, eter dietylowy lub diglym (CH
3−O−CH
2CH
2−O−CH
2CH
2−O−CH
3), w których monomeryczny boran BH
3 jest solwatowany przez cząsteczkę rozpuszczalnika tworząc układ kwas–zasada (według teorii Lewisa)[1][2].

- Kompleks boran–eter

Mechanizm reakcji
edytujBorowodór BH
3 jest pochodną boru, który – jak większość związków pierwiastków 13 grupy układu okresowego – posiada lukę elektronową, tzn. brakuje mu pary elektronowej do uzyskania oktetu elektronowego. Borowodór jest więc kwasem Lewisa, gdyż jest zdolny przyjąć parę elektronową na pusty orbital p.
Addycja borowodoru do alkenu jest reakcją skoordynowaną. Proces zaczyna się od ataku chmury elektronowej wiązania π alkenu na atom boru. Następnie zaczyna wykształcać się wiązanie boru z jednym z winylowych atomów węgla, podczas gdy drugi atom węgla tworzy częściowe wiązanie z jednym z atomów wodoru boranu. W stanie przejściowym reagujące cząsteczki tworzą czteroczłonowy układ cykliczny, w którym na atomie węgla niepołączonym z borem obecny jest częściowy ładunek dodatni[6]. Regioselektywność reakcji uwarunkowana jest nie przez trwałość karbokationu, lecz przez efekt steryczny[7]. Atom boru borowodoru przyłącza się do mniej podstawionego atomu węgla, ze względu na brak zawady sterycznej. Ponieważ nie powstaje wolny karbokation, podczas reakcji nie dochodzi do przegrupowania, co pozwala otrzymać wyłącznie jeden produkt reakcji, a nie mieszaninę produktów[1].

- Mechanizm reakcji hydroborowania

Powstający monoalkiloboran reaguje z kolejną cząsteczką alkenu dając następnie dialkiloboran i ostatecznie trialkiloboran[1].
Hydroborowanie-utlenianie
edytujPowstające w procesie hydroborowania alkiloborany mogą zostać poddane reakcji utleniania nadtlenkiem wodoru H
2O
2 w środowisku zasadowym, tworząc alkohole. Sumarycznie reakcje te mogą być traktowane jako addycja wody do alkenu przebiegająca niezgodnie z regułą Markownikowa, tj. dająca alkohol o niższej rzędowości niż uwodnienie alkenu katalizowane kwasem[1][2][6].

- Hydroborowanie-utlenianie

Mechanizm hydroborowania-utleniania
edytujW drugim etapie hydroborowania-utleniania następuje atak nukleofilowy anionu nadtlenowodorkowego na atom boru alkilowej pochodnej borowodoru. Migracja grupy alkilowej na atom tlenu zachodzi z retencją konfiguracji na atomie węgla. Reakcja biegnie raczej przez trialkiloboran B(OR)
3 niż monoalkiloboran BH
2OR:

- Mechanizm hydroborowania-utleniania

- Animacja zachodzącej reakcji


Stereochemia reakcji
edytujPrzyłączenie cząsteczki wody do cyklicznego alkenu daje produkt podstawienia zgodny z addycją syn, przeciwnie niż ma to miejsce w syntezie alkoholi w reakcji hydroksyrtęciowania połączonego z odrtęciowaniem, gdzie zachodzi addycja anti. Taki przebieg reakcji tłumaczony jest tworzeniem się pericyklicznego stanu przejściowego, w którym następuje przeniesienie anionu wodorkowego[1][8][9].

- Addycja syn cząsteczki H
2O do cyklicznego alkenu

Przebieg reakcji zgodnie z syn-addycją został również potwierdzony przez zastosowanie deuterowanego diboranu w reakcji z trans-2-butenem. W reakcji tej teoretycznie możliwe są dwa produkty: jeden izomer erytro powstający w wyniku cis-hydratacji i drugi izomer treo w wyniku trans-hydratacji. Badania spektroskopowe NMR wykazały, że powstaje wyłącznie izomer erytro[10].

- syn (cis) addycja cząsteczki H
2O do łańcuchowego alkenu

Hydroborowanie-utlenianie alkinów
edytujReakcja hydroborowania może zachodzić również na alkinach. Jeżeli użyty zostanie alkin terminalny produktem będzie aldehyd, jeżeli wiązanie potrójne alkinu będzie wewnątrz łańcucha węglowego, produktem będzie odpowiedni keton. W wyniku reakcji hydroborowania na alkinach, borowodór atakowany jest przez parę elektronową jednego tylko wiązania π, przez co początkowo powstają enole, które są nietrwałe i szybko ulegają przekształceniu do odpowiedniego aldehydu bądź ketonu w wyniku tautomerii keto-enolowej. Addycja ta również zachodzi w ułożeniu syn[11].

- Hydroborowanie-utlenianie terminalnego alkinu

Reakcje ze związkami α-bromokarbonylowymi
edytujAlkiloborany reagują z α-bromopochodnymi ketonów i estrów kwasów karboksylowych. Podobnie jak w acetylooctanie etylu czy estrze kwasu malonowego, tak i w α-bromoketonach oraz estrach α-bromokwsów proton przy drugim atomie węgla (proton α) wykazuje wysoką kwasowość, czyli może zostać znacznie łatwiej oderwany pod działaniem zasady niż w przypadku typowych grup alkilowych. W syntezie tej jako zasadę stosuje się najczęściej 2,6-di-tert-butylofenolan potasu, a jako czynnik alkilujący B-alkilo-9-borabicyklo[3.3.1]nonan (B-alkilo-9-BBN). Zastosowanie zasady o tak dużej objętości przestrzennej uniemożliwia zachodzenie konkurencyjnej reakcji substytucji nukleofilowej SN2. Z kolei zastosowanie bicyklicznego związku boroorganicznego zapewnia otrzymanie produktu monoalkilowania. 9-BBN otrzymuje się w reakcji cyklicznego dienu (1,5-cyklooktadienu) z diboranem B
2H
6[12]:

- Synteza B-alkilo pochodnej borabicyklo[3.3.1]nonanu

Mechanizm borowodorowania α-bromopochodnych ketonów i estrów
edytujMechanizm reakcji polega na oderwaniu protonu α, a powstający stabilny karboanion (zasada Lewisa) atakuje parą elektronową alkiloboran (kwas Lewisa), tworząc nowe wiązanie węgiel–bor (C
α−B). Jednocześnie z utworzeniem tego wiązania ładunek ujemy zostaje przeniesiony na atom boru, co powoduje osłabienie wiązania węgiel–bor z grupą alkilową, dzięki czemu grupa ta migruje jako anion na atom węgla. Ostatecznie następuje atak pary elektronowej atomu tlenu grupy OH 2,6-di-tert-butylofenolu na atom boru dając związek typu 9-BBN-zasada oraz α-alkiloketon bądź α-alkiloester[10].

- Mechanizm reakcji alkilowania α-bromoacetonu przez borowodorowanie

Cechy reakcji hydroborowania
edytuj- Orientacja addycji niezgodna z regułą Markownikowa
- Stereochemia addycji syn
- Brak przegrupowania karbokationów
- Możliwość syntezy alkoholi, których nie można otrzymać innymi metodami takimi jak: addycja wody w obecności jonów H+, hydroksyrtęciowanie, czy też synteza z zastosowaniem związków Grignarda.
Przypisy
edytuj- ↑ a b c d e f Robert T. Morrison, Robert N. Boyd, Chemia Organiczna, t. 1, Warszawa: Wydawnictwo Naukowe PWN, 1997, s. 587–590, ISBN 83-01-04-166-8.
- ↑ a b c Przemysław Mastalerz, Chemia Organiczna, Warszawa: Państwowe Wydawnictwo Naukowe, 1986, s. 408–409, ISBN 83-01-03379-7.
- ↑ a b Brown i inni, Selective Conversion of Olefins into Organoboranes Through Competitive Hydroboration, Isomerization and Displacement Reactions, „Journal of Organic Chemistry”, 22, 1957, s. 1137–1138, DOI: 10.1021/jo01360a626 (ang.).
- ↑ Herbert C. Brown, Biographical [online], Nobelprize.org [dostęp 2019-01-08] (ang.).
- ↑ Herbert C. Brown, From little acorns to tall oaks – from boranes through organoboranes. Nobel Lecture 1979, Nobelprize.org, 8 grudnia 1979 [dostęp 2019-01-08] (ang.).
- ↑ a b Herbert C. Brown, Negishi Eiichi, Hydroboration. XXXII. Cyclic hydroboration of dienes with thexylborane, „Journal of the American Chemical Society”, 94, 1972, s. 3567–3572, DOI: 10.1021/ja00765a051.
- ↑ Alfred Hassner, Conrad Pillar, Stereochemistry of Hydroboration, „Journal of Organic Chemistry”, 27, 1962, s. 2914–2915, DOI: 10.1021/jo01055a501 (ang.).
- ↑ Herbert C. Brown, George Zweifel, Hydroboration. IX. The Hydroboration of Cyclic and Bicyclic Olefins–Stereochemistry of the Hydroboration Reaction, „Journal of the American Chemical Society”, 83, 1961, s. 2544–2551, DOI: 10.1021/ja01472a028 (ang.).
- ↑ Herbert C. Brown, George Zweifel, A stereospecific cis hydration of the double bond in cyclic derivatives, „Journal of the American Chemical Society”, 81, 1959, s. 247, DOI: 10.1021/ja01510a059 (ang.).
- ↑ a b Robert T. Morrison, Robert N. Boyd, Chemia Organiczna, t. 2, Warszawa: Wydawnictwo Naukowe PWN, 1997, s. 55–57, ISBN 83-01-04-166-8.
- ↑ Herbert C. Brown, S.K. Gupta, Catecholborane (1,3,2-benzodioxaorole) as a new, general monohydroboration reagent for alkynes. Convenient synthesis of alkeneboronic esters and acids from alkynes via hydroboration, „Journal of the American Chemical Society”, 94, 1972, s. 4370–4371, DOI: 10.1021/ja00767a072 (ang.).
- ↑ George W. Kabalka, Newell S. Bowman, Stereochemistry of the hydroboration reaction, „Journal of Organic Chemistry”, 38, 1973, s. 1607–1608, DOI: 10.1021/jo00948a035 (ang.).