Jonofory
Jonofory – organiczne związki chemiczne zdolne do transferu jonów przez fazę hydrofobową rozdzielającą dwie fazy wodne. Termin antybiotyki jonoforowe jest stosowany do tych jonoforów, które wykazują działanie bakteriobójcze. Właściwości antybiotyczne jonoforów wynikają z ich zdolności do transportu jonów przez błony lipidowe komórek lub organelli. Jony, które nie mogą swobodnie przemieszczać się przez dwuwarstwy lipidowe, są transportowane w postaci kompleksów, w których jonofor pełni rolę „gospodarza”, a jon rolę „gościa”[1][2].

b) jonofor tworzący kanał
Odkrycie jonoforów
edytuj
Pierwszym poznanym antybiotykiem z grupy jonoforów była gramicydyna S (z ang. Gramicidin Soviet). Została ona odkryta już w 1942 roku przez rosyjskich naukowców – Gieorgija Gausego i jego żonę Marię Brażnikową – badających bakterie z gatunku Bacillus brevis. Badacze zaobserwowali, że gramicydyna S silnie hamuje rozrost kolonii gronkowca złocistego[3][4]. Nie znali oni jednak mechanizmu tego działania. Kwestia ta wciąż nie jest w pełni wyjaśniona, pomimo że określono już strukturę związku[5][6]. Przyjmuje się, że powoduje ona rozładowanie gradientu stężeń jonów na zewnątrz i wewnątrz komórki bakteryjnej, powodując jej obumieranie[7][8]. W czasach Gausego i Breżnikowej znane były także inne antybiotyki zaliczane obecnie do jonoforowych, takie jak gramicydyna A, walinomycyna, naktyny, wirginiamycyna i kwas lasalowy, mimo iż nie znano jeszcze właściwego sposobu ich działania.
Odkrycia właściwości jonoforetycznych dokonał Berton Pressman w 1967 roku. Jako pierwszy zaobserwował on udział małych niebiałkowych cząsteczek w transporcie jonów przez błony biologiczne. Wraz z Cyrilem Moore’em wykazali oni, że działanie antybiotyczne walinomycyny jest związane z obecnością kationów potasu[9]. Badania wykazały, że walinomycyna powoduje rozprzęganie procesu fosforylacji oksydacyjnej poprzez wymianę jonów wodorowych ze środka mitochondrium na kationy potasu[10][11][12]. Początkowo Pressman i Moore zakładali, że walinomycyna wspomaga działanie pompy sodowo-potasowej. W tamtym czasie był to jedyny znany sposób transportu jonów przez błony biologiczne. Nie potrafili oni jednak wytłumaczyć, dlaczego proces wymiany jonów nie zachodzi w przypadku zamiany kationów potasu na sodu[13]. Dopiero w 1967 roku Pressman podał właściwy sposób działania walinomycyny oraz gramicydyny A. Opublikował on wraz z innymi współautorami pracę, w której po raz pierwszy użył terminów ionophores (jonofory) oraz ion-carrying antibiotics (przenośniki jonów będące antybiotykami)[14]. Wykazali oni, że walinomycyna jest zdolna do selektywnego kompleksowania i transportowania przez błony lipidowe kationów potasu, lecz nie sodu, co tłumaczy wcześniejsze niejasności. W tym samym roku opublikowane zostały wyniki badań rentgenostrukturalnych kompleksu nonaktyny z kationem potasu[15], a dwa lata później opisana została struktura kompleksu walinomycyny z kationem potasu, co ostatecznie potwierdziło możliwość kompleksowania kationów metali przez te antybiotyki[16]. W strukturach krystalicznych obu tych kompleksów kation potasu związany jest przez osiem wiązań koordynacyjnych od atomów tlenu, tworzących niemal idealny układ oktaedryczny.
W tym samym czasie, w 1967 roku, Charles Pedersen dokonał innego przełomowego odkrycia. Prowadził badania nad katalizatorami polimeryzacji poszukując otwartołańcuchowego ligandu kompleksującego jony VO2+
[17][18]. Celem Pedersena było otrzymanie bis[2-(o-hydroksyfenoksy)etylo]eteru. Przeprowadzona synteza doprowadziła go jednak do cyklicznego związku dibenzo-18-korona-6. Pedersen jako pierwszy zauważył, że ten cykliczny eter jest zdolny do kompleksowania kationów sodu pochodzących zarówno z wodorotlenku, jak i nieorganicznych soli tego metalu. W dalszych badaniach Charls Pedersen otrzymał serię cyklicznych eterów, którym nadał nazwę „etery koronowe”, a także wykazał ich zdolność do tworzenia kompleksów z kationami metali[19]. Za swoje badania otrzymał on wraz z Donaldem Cramem i Jean-Marie Lehnem Nagrodę Nobla w dziedzinie chemii w 1987 roku[18][20]. Eter 12-korona-4 był znany jeszcze przed odkryciem Pedersena, bo już od 1957 roku, jednak zdolności kompleksotwórcze tego związku nie zostały wcześniej zaobserwowane[21]. Odkrycie Pedersena zainspirowało naukowców na całym świecie do poszukiwania nowych syntetycznych makroligandów i jest uznawane za początek chemii supramolekularnej[22].
Klasyfikacja jonoforów
edytujJonofory mogą zostać sklasyfikowane według różnych kryteriów: pochodzenia, budowy chemicznej cząsteczek, metodę transportu jonów czy selektywności względem kompleksowanych jonów. Główny podział uwzględniający sposób otrzymania jonoforów pozwala sklasyfikować je na dwie zasadnicze grupy: jonofory pochodzenia naturalnego i jonofory syntetyczne. Ze względu na metodę transportu jonów można je podzielić na nośnikowe oraz tworzące kanały jonowe. Biorąc pod uwagę budowę chemiczną cząsteczek jonoforów można je podzielić na cykliczne lub otwartołańcuchowe, a także inne rodzaje np. jonofory karboksylowe, oligopeptydy, cyklopeptydy, cyklodepsydy, cyklodepsypeptydy i inne[1][2][23]. Ze względu na rodzaj kompleksowanych jonów wyróżnia się jonofory kompleksujące kationy jednowartościowe i dwuwartościowe. Ostatecznie jonofory można podzielić ze względu na kompleksowane jony: sodu, potasu, wapnia itd.
| Jonofory pochodzenia naturalnego | Jonofory pochodzenia syntetycznego | |||||
|---|---|---|---|---|---|---|
| Neutralne cykliczne | Otwarto łańcuchowe | Neutralne cykliczne | Otwarto łańcuchowe | |||
| Cyklopeptydy | Cyklodepsydy | Cyklodepsypeptydy | Karboksylowe | Neutralne niecykliczne | Etery koronowe Kryptandy Sferandy Karcerandy |
Podandy (polietylenogligole) Oligotetrahydropirany |
| przykłady: gramicydyna S antramanid |
przykład: naktyny |
przykłady: walinomycyna wirginiamycyna |
przykłady: monenzyna kwas lasalowy |
przykład: gramicydyny A, B i C |
przykłady: 18-korona-6 dibenzo-18-korona-6 |
przykłady: PEG polioksymetylen |
Jonofory pochodzenia naturalnego
edytuj
Jonofory naturalne są związkami produkowanymi głównie przez bakterie: Streptomyces spp., Enterococcus spp. oraz Bacillus spp. Pozyskuje się je również z grzybów rodzaju Fusarium.
Wyróżnić można dwie zasadnicze grupy jonoforów naturalnego pochodzenia: karboksylowe oraz neutralne, które mogą być zarówno cykliczne, jak i otwartołańcuchowe.
Jonofory karboksylowe
edytuj
Jonofory karboksylowe zwane też antybiotykami polieterowymi posiadają grupę karboksylową (−COOH) i transportują kationy w postaci soli obojętnych R−COO−
M+
, gdzie M to kation metalu lub jon wodorowy H+
. Jonofor (makrocykliczy ligand) jest więc anionem. Cząsteczki jonoforów karboksylowych, choć zaliczają się do „otwarto-łańcuchowych”, nie są w rzeczywistości cząsteczkami liniowymi. Obecność grupy karboksylowej na jednym końcu cząsteczki oraz grup hydroksylowych na drugim daje możliwość tworzenia się wewnątrzcząsteczkowych wiązań wodorowych, co sprawia, że przyjmuje ona „pseudo-cykliczny” kształt. Eterowe atomy tlenu stanowiące „układ polieterowy” lub „szkielet polieterowy”, skierowane są do wnętrza cząsteczki tworząc hydrofilową wnękę zdolną do skompleksowania kationu[1][2][24]. Jednym z najlepiej przebadanych, a zarazem najpowszechniej stosowanych antybiotyków polieterowych jest monenzyna A[25].
| Jonofor | Źródło izolacji |
|---|---|
| Jonomycyna | Streptomyces conglobatus[26] |
| Kalcymycyna | Streptomyces chartreusensis[27] |
| Kwas lasalowy | Streptomyces lasaliensis[28] |
| Monenzyna | Streptomyces cinnamonensis[29] |
| Nigerycyna | Streptomyces violaceoniger[30] |
| Salinomycyna | Streptomyces albus[31] |
Jonofory neutralne
edytuj
Jonofory neutralne mogą być zarówno cykliczne, jak i niecykliczne. Cząsteczki tych ligandów są elektrycznie obojętne (nie posiadają ładunku), dlatego też z kationami metali tworzą kompleksy obdarzone ładunkiem. Aniony mogą, lecz nie muszą być transportowane równolegle (w symporcie) odpowiednim kanałem jonowym w zależności od rodzaju transportu[1][2].
| Jonofor | Źródło izolacji |
|---|---|
| Beauverycyna | Beauveria bassiana[32] oraz grzyby rodzaju Fusarium[33] |
| Enniatyny | Grzyby rodzaju Fusarium[34] |
| Gramicydyny | Bacillus brevis[3] |
| Naktyny | różne szczepy Actinomyces[35] |
| Walinomycyna | Streptomyces fulvissimus[36] |
Jonofory syntetyczne
edytujJonofory kationowe
edytuj- Cykliczne
Etery koronowe to dwuwymiarowe cykliczne jonofory syntetyczne[37]. Etery koronowe, w których cząsteczce znajduje się trzeciorzędowy atom azotu z przyłączonym łańcuchem bocznym zawierającym dodatkowe miejsca koordynacyjne, np. jednostki etylenooksy (−CH
2CH
2O−), nazywa się eterami lariatowymi lub lassowymi[38][39] (ang. lariat – lasso). Jeżeli taki dodatkowy łańcuch boczny jest połączony z pierścieniem eteru koronowego dwoma końcami (przez dwa atomy azotu), to analogi te nazywa się kryptandami[40]. Są to tzw. trójwymiarowe analogi eterów koronowych. Z kolei sferandy zawierają co najmniej dwa dodatkowe łańcuchy przyłączone do szkieletu eteru koronowego[41]. Trójwymiarowe analogi eterów koronowych zawierające wyłącznie atomy azotu zamiast atomów tlenu to sepulchrandy[1]. Znane są również cykliczne jonofory syntetyczne zawierające pierścienie aromatyczne jak kaliksareny czy karcerandy[42][43]. Wymienione grupy syntetycznych jonoforów cyklicznych to zaledwie kilka spośród ogromnej liczby otrzymanych do tej pory.
-
 Eter koronowy
Eter koronowy -
 Eter lariatowy
Eter lariatowy -
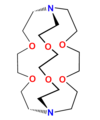 Kryptand
Kryptand -
 Sferand
Sferand -
 Sepulchrand
Sepulchrand





- Niecykliczne
Niecykliczne jonofory syntetyczne nazywane są ogólne podandami. Są to otwarto-łańcuchowe analogi eterów koronowych i podobnie jak one zawierają szereg heteroatomów (tlenu, azotu lub siarki), oddzielonych jednostkami etylenowymi −CH
2CH
2−. Zaliczają się do nich przede wszystkim polietylenoglikole (PEG) oraz oligotetrahydropirany. W przypadku tych drugich elektronodonorowe atomy są częścią niewielkiego pierścienia np. tetrahydrofuranowego[1][44].
-
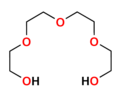 Polietylenoglikol
Polietylenoglikol -
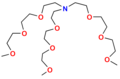 Tripodand
Tripodand -
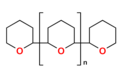 Oligotetrahydropiran
Oligotetrahydropiran



- Kompleksy jonoforów syntetycznych
Jonofory syntetyczne mogą tworzyć kompleksy o różnej stechiometrii w zależności od rozmiaru promienia jonowego kationu oraz rozmiaru makropierścienia (np. 1:1, 1:2, 2:1 jonofor:kation). Związki kompleksowe niektórych jonoforów syntetycznych posiadają osobne nazwy[45].
| Jonofor (ligand) | Kompleks |
|---|---|
| Karcerand | Karcerat |
| Kawitand | Kawitat |
| Koronand | Koronat |
| Kryptand | Kryptat |
| Podand | Podat |
| Sferand | Sferat |
-
 Kompleks 18-korona-6 z kationem K+
Kompleks 18-korona-6 z kationem K+
o stechiometrii 1:1 -
 Kompleks 12-korona-4 z kationem Li+
Kompleks 12-korona-4 z kationem Li+
o stechiometrii 2:1

lithium-cation-from-xtal-3D-balls-B.png)
Jonofory anionowe
edytujZnane są również syntetyczne jonofory dla anionów[45] m.in. CO2−
3, RCOO−
i OH−
, Cl−
, HSO−
3, NO−
2, SO2−
4.




Budowa cząsteczek
edytujCząsteczki jonoforów posiadają specyficzną budowę. Wewnętrzna część cząsteczki jest hydrofilowa, gdyż zawiera atomy pierwiastków o wysokiej elektroujemności głównie tlenu, azotu, rzadziej siarki. Obecna we wnętrzu cząsteczki hydrofilowa wnęka jest zdolna kompleksować kation metalu. Rozmiar wnęki ma zasadniczy wpływ na rodzaj jonów, jakie mogą być skompleksowane, czyli zasadniczo na selektywność procesu. Tylko jony o odpowiednim promieniu jonowym mogą zostać związane w hydrofilowej wnęce jonoforu.
Na selektywność kompleksowania wpływa także elastyczność konformacyjna cząsteczki oraz to, czy jest ona cykliczna, pseudo-cykliczna czy helikalna. Część zewnętrzną cząsteczki jonoforu stanowią najczęściej niepolarne grupy alkilowe (głównie metylowe, etylowe i izopropylowe), dlatego jest ona hydrofobowa. Jednocześnie niepolarny charakter grup alkilowych nadaje cząsteczce (zarówno samego jonoforu, jak i kompleksu) charakter lipofilowy, dzięki czemu mogą utrzymywać się w warstwie lipidowej[1][2]. Wyjątkiem są etery koronowe, które ze względu na giętkość konformacyjną mogą ulegać „wywinięciu” i stać się rozpuszczalne w rozpuszczalnikach polarnych, a nawet w wodzie. Rozpuszczalność eterów koronowych w wodzie jest jednak niska. Ulega ona znacznej poprawie w obecności wodorotlenków lub soli nieorganicznych w wyniku tworzenia kompleksów z dostępnymi kationami[19].
 |
 |

|
| Cząsteczka kompleksu monenzyny A z kationem Na+ . (jonofor naturalny) |
Cząsteczka kompleksu eteru 18-korona-6 z kationem K+ . (jonofor syntetyczny) |
Zmiana konformacji cząsteczki eteru koronowego w zależności od rozpuszczalnika |
Selektywność kompleksowania
edytujJonofory wykazują dużą selektywność kompleksowania jonów. Preferencja kompleksowania jonów przez jonofor zależy od jego trójwymiarowej struktury, różnicy pomiędzy energią desolwatacji i energią kompleksowania oraz stałej trwałości kompleksu. Selektywność jonoforu wynika z polarności hydrofilowej wnęki, lipofilowości zewnętrznej powierzchni, wiązań wodorowych obecnych w cząsteczce, a także oddziaływań typu jon-dipol pomiędzy kompleksowanym jonem a heteroatomami cząsteczki jonoforu. Duży wpływ na proces selektywnego rozpoznawania molekularnego ma również pH środowiska, siła jonowa roztworu oraz natura chemiczna rozpuszczalnika[45].
Selektywność kompleksowania kationów przez wybrane jonofory[2][45]
Kationy jednowartościowe:
- Kwas lasalowy: Cs+
> Rb+
≈ K+
> Na+
> Li+ - Monenzyna: Na+
> K+
≈ Li+
> Rb+
> Cs+ - Nigerycyna: K+
≈ Rb+
> Na+
> Cs+
> Li+ - Walinomycyna: Rb+
> K+
> Cs+
> Ag+
> NH+
4 > Na+
> Li+
Kationy dwuwartościowe:
Transport jonów
edytuj
- Jonofory typu nośnikowego
Jonofory nośnikowe kompleksują kation z fazy wodnej znajdując się na granicy faz, jednak pozostając w warstwie lipidowej. Kation jest następnie transportowany przez dwuwarstwę lipidową w postaci kompleksu. Po przeciwnej stronie błony, na granicy faz zachodzi dekompleksowanie kationu, po czym cząsteczka jonoforu wraca, aby wykonać kolejny cykl. Proces ten powtarza się do wyrównania stężeń jonu po obu stronach błony[11][12][14].
- Jonofory tworzące kanał
Kanały jonowe tworzone przez jonofory nie są trwałe, jak ma to miejsce w przypadku kanałów białkowych. Kanały jonoforowe powstają najczęściej z kilku cząsteczek jonoforu. Cząsteczki te ulegają samoorganizacji w taki sposób, aby powstał por, przez który jony mogą przedostać na drugą stronę błony. Kanały takie powstają tylko na krótki czas, po czym cząsteczki się rozdzielają, aż ponownie utworzą kanał z tymi samymi lub zupełnie innymi cząsteczkami. Przykładem jonoforu tworzącego kanał jest oligopeptyd – gramicydyna A. Kanał tworzony przez ten jonofor zbudowany jest z dwóch cząsteczek ułożonych względem siebie N-końcami peptydu „głowa do głowy”. Kanał gramicydyny jest zdolny transportować różne kationy jednowartościowe, lecz nie transportuje kationów dwuwartościowych ani anionów[46]. Ester metylowy monenzyny A (syntetyczna pochodna monenzyny) jest zdolny do tworzenia kanału protonowego w wyniku samoorganizacji aż ośmiu cząsteczek[47].
- Rodzaje transportu jonów
Wyróżnia się kilka sposobów transportu jonów przez błony lipidowe z udziałem jonoforów. Jeżeli w wyniku procesu potencjał transbłonowy ulega zmianie, proces nosi nazwę transportu elektrogenicznego. Jeżeli różnica potencjałów po obu stronach błony zostaje zachowana, transport taki nazywa się elektroneutralnym.
Jonofory neutralne dokonują najczęściej transportu elektrogenicznego, gdyż transportują konkretny rodzaj kationów w jedną stronę, co wywołuje zmianę potencjałów[2][48]. Jonofory karboksylowe transportują w jedną stronę błony kation metalu w postaci soli (karboksylanu) −COO−
M+
, a w przeciwną jon wodorowy w postaci grupy karboksylowej −COOH. Jest to zatem transport elektroneutralny, który został nazwany dla tej grupy jonoforów transportem biologicznym[2]. Wymianę jonów przez jonofory karboksylowe zapisuje się często w następujący sposób: M+
/H+
[49]. Jonofory karboksylowe po modyfikacji grupy karboksylowej np. na estrową czy amidową, również mogą dokonywać transportu jonów i również wykazują właściwości przeciwdrobnoustrojowe. Modyfikowane jonofory karboksylowe transportują jeden rodzaj jonów w jednym kierunku, a inny rodzaj jonów w przeciwnym. Transport taki został nazwany transportem biomimetycznym od greckich słów „bios” – życie – i „mimesis” – naśladować – a zapisuje się go M+
/N+
, gdzie M+
i N+
to kationy metali[50][51].
Niektóre jonofory mogą transportować jony przez błony lipidowe jednocześnie na sposób elektroneutralny, jak i elektrogeniczny, np. monenzyna A[52]. W takim przypadku mówi się o transporcie mieszanym[53]. Sposoby transportu jonów przez poszczególne jonofory są wciąż kwestią dyskusyjną i nie są w pełni wyjaśnione.
Właściwości przeciwdrobnoustrojowe
edytujJonofory to liczna grupa związków organicznych, zarówno naturalnych, jak i syntetycznych. Większość z nich wykazuje działanie przeciwdrobnoustrojowe. Od czasu ich odkrycia udowodniono, że mogą one wykazywać działanie przeciwbakteryjne, przeciwwirusowe, przeciwgrzybicze, przeciwnowotworowe, przeciwmalaryczne czy kokcydiostatyczne[2][3][7][54].
Mechanizm działania antybiotycznego
edytujMechanizm działania antybiotycznego jonoforów jest ściśle związany z ich zdolnością do transportowania jonów przez błony biologiczne komórek oraz organelli komórkowych, w szczególności mitochondriów. Stężenia poszczególnych jonów po obu stronach błon są różne. Naturalny gradient stężenia jonów: Na+
, K+
, Ca2+
, Mg2+
, a także jonów wodorowych H+
(czyli różnicy pH) jest niezbędny do prawidłowego funkcjonowania białkowych kanałów jonowych. Rozpuszczalne w błonach fosfolipidowych cząsteczki jonoforów powodują jego zniesienie, co zaburza fosforylację oksydacyjną i w konsekwencji prowadzi do śmierci komórek[3][4][11][13][51]. Wiele jonoforów wykazuje działanie bakteriobójcze selektywnie wobec bakterii Gram-dodatnich. Wynika to najprawdopodobniej z tego, że u bakterii Gram-ujemnych ściana komórkowa zawiera dodatkowo warstwę peptydoglikanu, który utrudnia penetrację błon lipidowych[54][55].
Zastosowanie
edytujJonofory mają bardzo wiele zastosowań w zależności od rodzaju kompleksowanych jonów, selektywności, sposobu transportu przez błony oraz wykazywanego działania bakteriobójczego. Ze względu na wysoką toksyczność (np. walinomycyna czy ciguatoksyna) mają ograniczoną przydatność w farmakoterapii[56][57].
- Przykładowe zastosowania jonoforów
- Jonofory karboksylowe takie jak np. monenzyna, kwas lasalowy czy salinomycyna znalazły zastosowanie jako kokcydiostatyki oraz niehormonalne stymulatory wzrostu w przemysłowej hodowli drobiu lub bydła[56].
- Z kolei niektóre cyklopeptydy jak kapreomycyna, wiomycyna czy polimyksyny są niekiedy stosowane w leczeniu gruźlicy. Cykliczne oligopeptydy są stosowane powszechnie jako składniki maści stosowanych w leczeniu infekcji skórnych wywołanych przez ziarenkowce Gram-dodatnie[56].
- Depsypeptydy należące do streptogramin B, m.in. wirginiamycyna czy prystynamycyna, są stosowane pozajelitowo przy zakażeniach wywołanych przez wielooporne bakterie Gram-dodatnie oraz enterokoki[56].
- Zarówno jonofory pochodzenia naturalnego, jak i syntetyczne (szczególnie etery koronowe) są stosowane powszechnie w jonoselektywnych elektrodach (ISE) do wykrywania poszczególnych jonów.
- Podandy są coraz powszechniej stosowane jako rozpuszczalniki w syntezie organicznej. Wiele z nich (np. polietylenoglikole – PEGi), jest cieczami. Dzięki swojej naturze chemicznej mają dużą zdolność solwatacji jonów. Ta cecha jest szczególnie istotna, gdy wymagana jest stabilizacja produktów pośrednich jakimi mogą być karbokationy czy karboaniony. Rozpuszczalniki podandowe mogą być też stosowane jako mieszaniny z tradycyjnymi rozpuszczalnikami organicznymi[58].
- Etery koronowe są stosowane w katalizie przeniesienia międzyfazowego ze względu na ich zdolność zwiększania stopnia rozpuszczalności kationów nieorganicznych w fazie hydrofobowej[59][60].
Zobacz też
edytujPrzypisy
edytuj- ↑ a b c d e f g R. Ferdani, G.W. Gokel: Ionophores. W: Encyclopedia of supramolecular chemistry. s. 1401–1411.
- ↑ a b c d e f g h i Berton Pressman. Biological applications of ionophores. „Annu. Rev. Biochem.”. 45, s. 501–530, 1976. DOI: 10.1146/annurev.bi.45.070176.002441. (ang.).
- ↑ a b c d G.F. Gause, M.G. Brazhnikova. Gramicidin S and its use in the treatment of infected wounds. „Nature”. 154, s. 703, 1944. DOI: 10.1038/154703a0. (ang.).
- ↑ a b G.F. Gause, M.G. Brazhnikova. Gramicidin S origin and mode of action. „The Lancet”. 244, s. 715–716, 1944. DOI: 10.1016/S0140-6736(00)88377-4. (ang.).
- ↑ F. Sanger. The free amino group of gramicidin S. „Biochem. J.”. 40, s. 261–262, 1946. PMCID: PMC1258332. (ang.).
- ↑ R. Consden, A.H. Gordon, A.J.P. Martin, R.L.M. Synge. Gramicidin S: the sequence of the amino-acid residues. „Biochem. J.”. 41, s. 596–602, 1947. PMCID: PMC1258544. (ang.).
- ↑ a b N. Izumiya, T. Kato, H. Aoyaga, M. Waki, M. Kondo: Synthetic Aspects of Biologically Active Cyclic Peptides: Gramicidin S and Tyrocidines. New York: Wiley, 1979. ISBN 0-470-26863-8.
- ↑ E.J. Prenner i inni. Structure-Activity Relationships of Diastereomeric Lysine Ring Size Analogs of the Antimicrobial Peptide Gramicidin S: Mechanism of action and discrimination between bacterial and animal cell membranes. „J. Biol. Chem.”. 280, s. 2002–2011, 2005. DOI: 10.1074/jbc.M406509200. (ang.).
- ↑ C. Moore, B.C. Pressman. Mechanism of action of valinomycin on mitochondria. „Biochem. Biophys. Res. Commun.”. 15, s. 562–567, 1964. DOI: 10.1016/0006-291X(64)90505-4. (ang.).
- ↑ M. Hofert, B.C. Pressman. Stimulation of oxidative phosphorylation in mitochondria by potassium in the presence of valinomycin. „Biochemistry”. 5, s. 3919–3925, 1965. DOI: 10.1021/bi00876a025. (ang.).
- ↑ a b c B.C. Pressman. Inducted active transport of ions in mitochondria. „PNAS”. 3, s. 1076–1083, 1965. (ang.).
- ↑ a b L. Ernstern, G. Schatz. Mitochondria: A Historical Review. „J. Cell Biol.”. 91, s. 227–255, 1981. (ang.).
- ↑ a b E. Ogata, H. Rasmussen. Valinomycin and mitochondrial ion transport. „Biochemistry”. 5, s. 57–66, 1966. DOI: 10.1021/bi00865a009. (ang.).
- ↑ a b B.C. Pressman, E.J. Harris, W.S. Jagger, J.H. Johnson. Antibiotic-mediated transport of alkali ions across lipid barriers. „PNAS”. 58, s. 1949–1956, 1967. DOI: 10.1073/pnas.58.5.1949. (ang.).
- ↑ B.T. Kilbourn, J.D. Dunitz, L.A.R. Pioda, W. Simon. Structure of the K+ complex with nonactin, a macrotetrolide antibiotic possessing highly specific K+ transport properties. „J. Mol. Biol.”. 30, s. 559–563, 1967. DOI: 10.1016/0022-2836(67)90370-1. (ang.).
- ↑ M. Pinkerton, L.K. Steinrauf, P. Dawkins. The molecular structure and some transport properties of valinomcin. „Biochem. Biophys. Res. Commun.”. 35, s. 512–518, 1969. DOI: 10.1016/0006-291X(69)90376-3. (ang.).
- ↑ C.J. Pedersen, Chelated compounds of vanadium and substituted phenols US-Patent 3361778 Du Pont, United States Patent Office, 1968 (ang.).
- ↑ a b C.J. Pedersen. The discovery of crown ether (Noble Lecture). „Angew. Chem. Int. Ed. Eng.”. 27, s. 1021–1027, 1988. DOI: 10.1002/anie.198810211. (ang.).
- ↑ a b C.J. Pedersen. Cyclic polyethers and their complexes with metal salts. „J. Am. Chem. Soc.”. 89, s. 7017–7036, 1967. DOI: 10.1021/ja01002a035. (ang.).
- ↑ Charles J. Pedersen: The discovery of crown ethers Lobel lecture. 1987-12-08. [dostęp 2019-01-14]. (ang.).
- ↑ D.G. Stewart, D.Y. Waddan, E.T. Borrows, Improvements in and relating to the production of alkylene oxide polymers, United Kingdom Patent GB785229, 23 października 1957.
- ↑ J.W. Steed: Definition. W: Encyclopedia of supramolecular chemistry. s. 1401–1411.
- ↑ M. Dobler: Natural Cation-binding Agents. W: G.W. Gokel: Molecular Recognition: Receptors for Cationic Guests. Pergamon, 1996, s. 267–313. ISBN 0-08-042713-8.
- ↑ B.C. Pressman, N.T. DeGuzman. Biological applications of ionophores: theory and practice. „Ann N Y Acad Sci.”. 264, s. 373–386, 1975. DOI: 10.1111/j.1749-6632.1975.tb31497.x. (ang.).
- ↑ J.W. Westley, Polyether antibiotics: Versatile carboxylic acid ionophores produced by Streptomyces, D. Perlman (red.), „Advances in Applied Microbiology”, 22, New York, San Francisco, London: Academic Press, 1977, s. 177–223, DOI: 10.1016/S0065-2164(08)70163-1. ISBN 978-0-12-002622-7.
- ↑ W.-C. Liu i inni. Ionomycin, a new polyether antibiotic. „J. Antibiot.”. 31, s. 815–819, 1978. DOI: 10.7164/antibiotics.31.815. (ang.).
- ↑ R.M. Gale, C.E. Higgins, M.M. Hoehn, Antibiotic A23187 and Process for preparation thereof, US Patent 3923823, 1975.
- ↑ J. Berger, A.I. Rachlin, W.E. Scott, L.H. Sternbach i inni. The Isolation of Three New Crystalline Antibiotics from Streptomyces. „J. Am. Chem. Soc.”. 73, s. 5295–5298, 1951. DOI: 10.1021/ja01155a084. (ang.).
- ↑ M.E. Haney Jr., M.M. Hoehn. Monensin, a new biologically active compound. I. Discovery and isolation. „Antimicrob. Agents Chemother.”. 7, s. 349–352, 1967. PMID: 5596158. (ang.).
- ↑ R.L. Harned i inni. Nigericin, a new crystalline antibiotic from an unidentified streptomyces. „Antibiot. Chemother.”. 1, s. 594, 1951. PMID: 24541690. (ang.).
- ↑ M. Mitani, T. Yamanishi, Y. Miyazaki. Salinomycin: A new monovalent cation ionophore. „Biochem. Biophys. Res. Commun.”. 66, s. 1231–1236, 1975. DOI: 10.1016/0006-291X(75)90490-8. (ang.).
- ↑ F.R. Champlin, E.A. Grula. Noninvolvement of Beauvericin in the entomopathogenicity of Beauveria bassiana. „Appl. Environ. Microbiol.”. 37, s. 1122–1126, 1979. PMID: 573587. PMCID: PMC243365. (ang.).
- ↑ A. Logrieco, A. Moretti, G. Castella. Beauvericin production by Fusarium species. „Appl. Environ. Microbiol.”. 64, s. 3084–3088, 1998. PMID: 9687479. PMCID: PMC106821. (ang.).
- ↑ E. Gäumann, Stephi Roth i inni. Enniatin, ein neues, gegen Mykobakterien wirksames Antibiotikum. „Experientia”. 3, s. 202–203, 1947. DOI: 10.1007/BF02163993. (niem.).
- ↑ R. Corbaz i inni. Stoffwechselprodukte von Actinomyceten. 3. Mitteilung. Nonactin. „Helv. Chim. Acta”. 38, s. 1445–1448, 1955. DOI: 10.1002/hlca.19550380617. (niem.).
- ↑ H. Brockmann, G. Schmidt-Kastner. Valinomycin I, XXVII. Mitteil. über antibiotica aus actinomyceten. „Chem. Ber.”. 88, s. 57–61, 1955. DOI: 10.1002/cber.19550880111. (niem.).
- ↑ G.W. Gokel: Crown ethers. W: Encyclopedia of supramolecular chemistry. s. 326–333.
- ↑ Lariat ethers. W: G.W. Gokel: Encyclopedia of supramolecular chemistry. s. 728.
- ↑ lariat ethers, [w:] A.D. McNaught, A. Wilkinson, Compendium of Chemical Terminology (Gold Book), S.J. Chalk (akt.), International Union of Pure and Applied Chemistry, wyd. 2, Oxford: Blackwell Scientific Publications, 1997, DOI: 10.1351/goldbook.L03458, ISBN 0-9678550-9-8 (ang.).
- ↑ B. Dietrich: Cryptands. W: Encyclopedia of supramolecular chemistry. s. 334–339.
- ↑ J.C. Sherman: Spherands. W: Encyclopedia of supramolecular chemistry. s. 1344–1348.
- ↑ I. Stihor, P. Lhotak: Calixarenes and their Analogues: Molecular Comlexation. W: Encyclopedia of supramolecular chemistry. s. 145–153.
- ↑ B.C. Cihh: Carcerands and Hemicarcerands. W: Encyclopedia of supramolecular chemistry. s. 189.
- ↑ E. Weber: Podands. W: Encyclopedia of supramolecular chemistry. s. 1106–1119.
- ↑ a b c d e G. Schroeder, B. Gierczyk: 1. Syntetyczne receptory jonowe – jonofory. W: Syntetyczne receptory jonowe. Poznań: Betagraf P.U.H., 2005. ISBN 83-89936-05-4. [dostęp 2014-05-25]. Wersja on-line: Wielkopolska Biblioteka Cyfrowa.
- ↑ D.W. Urry. The Gramicidin A Transmembrane Channel: A Proposed π(L,D) Helix. „PNAS”. 68, s. 672–676, 1971. PMID: 5276779. PMCID: PMC389014. (ang.).
- ↑ A. Huczyński, P. Przybylski, B. Brzezinski, F. Bartl. Spectroscopic and semiempirical studies of a proton channel formed by the methyl ester of Monensin A. „J. Phys. Chem. B”. 110, s. 15615–15623, 2006. DOI: 10.1021/jp062160o. (ang.).
- ↑ D.C. Tosteson, B.F. Gisin. Ion transport mediated by the valinomycin analogue cyclo(LLac- L-Val-D-Pro-D-Val)3 in lipid bilayer membranes. „J. Gen. Physiol.”. 77, s. 387–417, 1981. DOI: 10.1085/jgp.77.4.387. PMID: 7241088. PMCID: PMC2215419. (ang.).
- ↑ Y. Itoh, M.J. Law, L. Sokoloff. Effects of the Na+/H+ exchanger monensin on intracellular pH in astroglia. „Brain Research”. 882, s. 226–229, 2000. DOI: 10.1016/S0006-8993(00)02822-5. (ang.).
- ↑ H. Tsukube i inni. Biomimetic transport of unusual metal cations and amino acid ester salts mediated by monensin and lasalocid A. „J. Chem. Soc., Chem. Commun.”, s. 448–449, 1986. DOI: 10.1039/C39860000448. (ang.).
- ↑ a b H. Tsukube i inni. Biomimetic membrane transport: Interesting ionophores functions of naturally occurring polyether antibiotics toward unusual metal cations and amino did ester salts. „Inorg. Chem.”. 33, s. 2984–2987, 1994. DOI: 10.1021/ic00091a043. (ang.).
- ↑ A. Huczyński, J. Jaczkak, D. Łowicki, B. Brzezinski. Monensin A acid complexes as a model of electrogenic transport of sodium cation. „Biochim. Biophys. Acta”. 1818, s. 2108–2119, 2012. DOI: 10.1016/j.bbamem.2012.04.017.
- ↑ M. Inabayashi, S. Miyauchi, N. Kamo, T. Jin. Conductance change in phospholipid bilayer membrane by an electroneutral ionophore, monensin. „Biochemistry”. 34, s. 3455–3460, 1995. DOI: 10.1021/bi00010a038. (ang.).
- ↑ a b J. Stefańska, A. Huczyński. Biologiczne właściwości monenzyny A. „Biulety Wydz. Farmacji UMW”. 2, s. 1–12, 2008. (pol.).
- ↑ A. Huczyński, J. Stefańska i inni. Synthesis and antimicrobial properties of Monensin A esters. „Bioorg. Med. Chem. Lett.”. 18, s. 2585–2589, 2008. DOI: 10.1016/j.bmcl.2008.03.038. (ang.).
- ↑ a b c d Antybiotyki peptydowe. W: A. Chmiel, S. Grudziński: Biotechnologia i chemia antybiotyków. Warszawa: Wydawnictwo Naukowe PWN, 1998, s. 252–273. ISBN 83-01-12787-2.
- ↑ Satake M.,Murata M.,Yasumoto T.. The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus toxicus. „Tetrahedron Lett.”. 34, s. 1975–1978, 1993. DOI: 10.1016/S0040-4039(00)91978-6. (ang.).
- ↑ G. Schroeder, B. Łęska: Rozpuszczalniki podandowe. W: Kompleksy typu gość-gospodarz. Poznań: Betagraf P.U.H., 2003, s. 18–39. ISBN 83-918771-1-6. [dostęp 2014-05-25]. Wersja on-line: Wielkopolska Biblioteka Cyfrowa.
- ↑ R. Narayan, G.T. Tsao. Application of crown ethers as phase transfer catalysts in the electron transfer reactions of coal. „Conference: 186. national meeting of the American Chemical Society, Washington, DC, USA, 28 Aug 1983”. s. 261–267. (ang.).
- ↑ 18-Crown-6. W: C.L. Liotta, J. Berknerin: Encyclopedia of Reagents for Organic Synthesis. L. Paquette (red.). New York: J. Wiley & Sons, 2001. DOI: 10.1002/047084289X.rc261. ISBN 978-0-470-84289-8.
Bibliografia
edytuj- Encyclopedia of Supramolecular Chemisty. J.L. Atwood, J.W. Steed (redaktorzy). Boca Raton: CRC Pres Taylor & Francis Group, 2004. ISBN 0-8247-4723-2.
- Structures and Properties of Naturally Occurring Polyether Antibiotics, J. Rutkowski, B. Brzezinski; artykuł przeglądowy w języku angielskim
- Structure and Antimicrobial Properties of Monensin A and Its Derivatives: Summary of the Achievements, D. Łowkcki, A. Huczyński; artykuł przeglądowy w języku angielskim